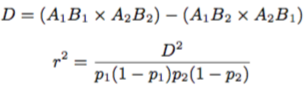Summary Statistic Files
Summary_Statistics_DataFrame_Performance:
- This provides summary statistics on the phenotypic and genetic trend across generations. If only one quantitative trait is simulated, columns for trait 2 are not displayed.
Generation: Generation number.
phen1: Mean (variance) phenotypic value for trait 1.
ebv1: Mean (variance) estimated breeding value for trait 1.
tgv1: Mean (variance) true genotypic breeding value for trait 1.
tbv1: Mean (variance) true breeding breeding value for trait 1.
tdd1: Mean (variance) true dominance deviation for trait 1.
res1: Mean (variance) residual value for trait 1.
phen2: Mean (variance) phenotypic value for trait 2.
ebv2: Mean (variance) estimated breeding value for trait 2.
tgv2: Mean (variance) true genotypic breeding value for trait 2.
tbv2: Mean (variance) true breeding breeding value for trait 2.
tdd2: Mean (variance) true dominance deviation for trait 2.
res2: Mean (variance) residual value for trait 2.
index tbv: Mean (variance) index true breeding value.
Summary_Statistics_DataFrame_Inbreeding:
- This provides summary statistics on the inbreeding and fitness trends across generations.
Generation: Generation number.
ped_f: Mean pedigree based inbreeding parameter.
gen_f: Mean genomic relationship diagonal constructed based on Van Raden (2008).
h1_f: Mean diagonal of haplotype based relationship matrix (Hickey et al. 2012; H1).
h2_f: Mean diagonal of haplotype based relationship matrix (Hickey et al. 2012; H2).
h3_f: Mean diagonal of ROH based relationship matrix (Howard et al. 2017).
homozy: Mean proportion homozygous (i.e. 1 - homozy = Observed Heterozygosity).
PropROH: Mean proportion of the genome in ROH of a given length.
ExpHet: Expected Heterozygosity (i.e. Σ (1 - p2 - q2)).
fitness: Mean multiplicative fitness value of an individual.
homozlethal: Mean number of homozygous FTL classified as lethal.
hetezlethal: Mean number of heterozygous FTL classified as lethal.
homozysublethal: Mean number of homozygous FTL classified as sub-lethal.
hetezsublethal: Mean number of heterozygous FTL classified as sub-lethal.
lethalequiv: Mean lethal equivalents (Lethal equivalents = Σ s for an animal).
Summary_Statistics_QTL:
- This provides summary statistics on the number of QTL/FTL segregating across generations.
Generation: Generation number.
Quant Founder Start: Number of QTL from founder generation segregating.
Quant Founder Lost: Number of QTL from founder generation fixed.
Mutation Quan Total: Number of QTL from new mutations segregating.
Mutation Quan Lost: Number of QTL from new mutations fixed.
Additive Var: True additive genetic variance based on Σ 2pq[a+d(q-p)]2.
Dominance Var: True dominance genetic variance based on Σ (2pqd)2.
Fit Founder Start: Number of FTL from founder generation segregating.
Fit Founder Lost: Number of FTL derived from founder generation fixed.
Mutation Fit Total: Number of FTL derived from new mutations segregating.
Mutation Fit Lost: Number of FTL derived from new mutations fixed.
Avg Haplotypes Window: Mean haplotypes contained within a haplotype window.
ProgenyDiedFitness: Number of progeny that died due to fitness.
LD_Decay:
- A file that has the average correlation (r
2) between two SNP across a range of distances. The average was generated by moving across the genome in 10 Mb blocks
and randomly grabbing two SNP and calculating their respective (r
2) and placing them in the correct bins based how far they were apart. Within a block 500 pairs of
SNP are randomly sampled and once finished the window is shifted by 5 Mb and is repeated until the end of the chromosome. This is conducted within each chromosome. The
distances are in the first row and are in Kilobases. Each row after the first row corresponds to the generation, such that line 2 is generation 0, line 3 is generation 1, etc.
The formula to calculate the D and (r2) values is below and the subscript refers to either SNP marker 1 or 2.
QTL_LD_Decay:
- Similar to the "LD Decay" file this file estimates the average correlation between a SNP and a QTL within window sizes of 0-0.5 Mb, 0.5-1.0 Mb, 1.0-1.5 Mb, 1.5-2.0 Mb and 2.0-2.5 Mb.
For example if a SNP and QTL were 0.75 Mb apart it would get placed in the 0.5 - 1.0 Mb bin. Each line in the file represents a QTL. If the correlation could not be estimated due to
lack of SNP in the window or if SNP are near fixation a ’-5’ will be produced. The first column represents the chromosome and the second column represents the position in Mb. The last
column is the average correlation within a generation across the different window sizes. Correlations within a generation across windows are separated by a ":" and sets of correlations
across generations are seperated by a "_".
Phase_Persistance_Generation:
- Using the output from the Phase Persistance file, the correlation between phases across generations was estimated within window sizes of 0-0.5 Mb, 0.5-1.0 Mb, 1.0-1.5 Mb, 1.5-2.0 Mb and
2.0-2.5 Mb. The first column is the generation and the columns following would be the correlation between the current generation and preceding generations.
TrainReference:
- A file with the estimated breeding value (EBV) and true breeding value (TBV). The output can be used to generate the accuracy and bias of EBV across generations.
ID: ID of individual.
T1_EBV: Estimated breeding value for trait 1.
T1_TBV: True breeding value for trait 1.
T2_EBV: Estimated breeding value for trait 2.
T2_TBV: True breeding value for trait 2.
Generation: Generation EBV was calculated.
Group: Whether animal was a parent or a selection candidate.
AmaxGeneration:
- This file estimates the mean maximum relationship for individuals that were born in a previous generation with selection candidates.
Summary_Statistics_ROH_Freq:
- The first two columns are the chromosomal and nucleotide position of the SNP and the remaining columns are the frequency of that SNP being in an ROH of the length
that was specified for a given generation. A SNP may not be in a window of a given length and therefore is set to -5.
Summary_Statistics_ROH_Length:
- The first two columns are the chromosomal and nucleotide position of the SNP and the remaining columns are the mean and median length (i.e. ”Mean_Median”) of ROH
for that given SNP when that SNP is in a ROH for a given generation. A SNP may not be in a window of a given length and therefore is set to -5.
WindowAdditiveVariance:
- Provides the true additive genetic variance for a given 1-Mb window across generations. The first row is the chromosome and mid-point in the window and the
remaining lines are the associated additive genetic variance estimates for a given window.
WindowDominanceVariance:
- Provides the true dominance genetic variance for a given 1-Mb window across generations. The first row is the chromosome and mid-point in the window and the
remaining lines are the associated dominance genetic variance estimates for a given window.